

学术前沿
今天小仲要分享的文章可不得了!哈尔滨医科大学团队对国自然热点“铁死亡”进行深入的剖析,探讨了“铁死亡”在心肌损伤和心功能障碍中的潜在作用和机制。在本研究中,研究者采用了一种多维度的研究方法,包括动物模型实验、RNA测序分析、多种病理染色、蛋白质组芯片、分子对接和动力学模拟等(有你感兴趣的吗?如果有,你可以来寻找精通各种研究方法的小仲的帮助哦!)。现在,就来看一看这篇文章都有哪些亮点值得反复咀嚼吧!
1、文章采用了多种实验方法和技术,包括动物模型实验、多种病理染色、分子对接和动力学模拟,这些方法的结合使得研究结果更加可靠和有说服力。
2、结合RNA-Seq和蛋白质组芯片分析确定PrA的分子靶点,为铁死亡的分子机制提供了多维度的理解。
3、文章深入探讨了国自然热点“铁死亡”,贴近热点,无疑让文章在思路上更甚一筹!
对于高质量的文章,严谨创新的研究思路和前沿高端的研究方法缺一不可!正如今天的这一篇文章,实验方法和技术简直让人眼前一亮!如果你也想掌握如此多的研究方法,也想拥有属于你的研究成果,就快行动起来吧!小仲这里有很多优秀新颖的研究思路可以帮助你,你在研究中有任何困难,也都可以来找小仲哦!

题目:原皂苷A通过靶向ACSL4/FTH1轴依赖性铁死亡保护DOX诱导的心肌损伤和心功能障碍
杂志:Advanced Science
影响因子:IF=14.3
发表时间:2024年7月
公众号回复“123”领取原文PDF,文献编号:20240803
研究背景
铁死亡是一种新的细胞死亡过程,主要表现为线粒体疾病、铁代谢异常、脂质过度过氧化和谷胱甘肽过氧化物酶4(GPX4)/谷胱甘肽(GSH)轴不平衡。现有证据表明,铁死亡与DOX介导的心功能障碍密切相关。最近,在DOX诱导的心肌细胞中发现铁嗜性表型。基于上述发现,开发针对上铁死亡的靶向药物有可能减轻DOX引起的心肌损伤。
原苏木苷A(PrA)是一种从苏木精中提取的活性成分,具有多种药理作用,包括抗氧化、抗炎和抗凋亡活性。前期研究表明,PrA可通过NF-kB和Akt/mTOR对心脏移植和自身免疫性心肌炎发挥免疫抑制作用。但PrA在DIC中的作用及其与铁死亡的关系仍不清楚。因此,本研究旨在探讨PrA对DIC的影响及其机制。
研究思路
本文首先建立DOX诱导的心肌病小鼠模型,给予PrA和DXZ(铁螯合剂)进行治疗发现PrA的心脏保护作用。接下来通过RNA测序分析和GSEA发现PrA对铁死亡的抑制作用。最后,研究人员利用HuProt人类蛋白质微阵列,结合生物素标记的PrA(Bio-PrA)和Cy5偶联链霉亲和素(Cy5-SA),筛选PrA的潜在靶标蛋白,并通过分子对接和动力学模拟进一步验证相互作用的分子细节,得出PrA通过靶向ACSL4/FTH1轴抑制铁死亡
研究结果
1.PrA减轻DOX诱导的心肌损伤和心功能障碍
为研究PrA在DOX诱导的心脏毒性中的作用,研究人员建立DIC小鼠模型,并在指定天数内用不同浓度的PrA或DXZ处理(图1A,B)。结果显示,与DXZ类似,PrA显著提高DOX处理小鼠的存活率,且呈剂量依赖性(图1C)。超声心动图评估显示,与对照组相比,DIC小鼠的心功能明显下降,主要体现在左室射血分数(LVEF)、左室缩短分数(FS)、左室舒张内径(LVIDd)和左室收缩内径(LVIDs)参数上。相比之下,用DXZ或增加剂量的PrA治疗后,心功能部分恢复,表明PrA对DOX诱导的心功能障碍有保护作用(图1D-H)。通过分析心肌损伤生物标志物和心脏组织病理学变化来评估PrA对DOX诱导的心肌损伤的影响。生化分析显示,PrA治疗显著抑制DOX诱导的血清肌酸激酶-MB(CK-MB)和乳酸脱氢酶(LDH)水平升高且呈剂量-反应关系(图1I,J)。苏木精和伊红(H&E)染色显示DIC小鼠有严重的炎症浸润,通过增加PrA浓度可减轻炎症浸润(图1K)。此外,通过小麦胚芽凝集素(WGA)、Sirius Red和Masson’s三色染色,PrA剂量依赖性地抑制DOX诱导的小鼠心肌细胞肥大和心肌纤维化(图1L-Q)。TUNEL染色证实,PrA处理可改善DOX诱导的心肌DNA损伤(图1R,S)。值得注意的是,高浓度PrA在DIC中表现出心脏保护作用(图1I,J)。上述数据表明,PrA可减轻DOX诱导的心脏损伤并预防心功能障碍。
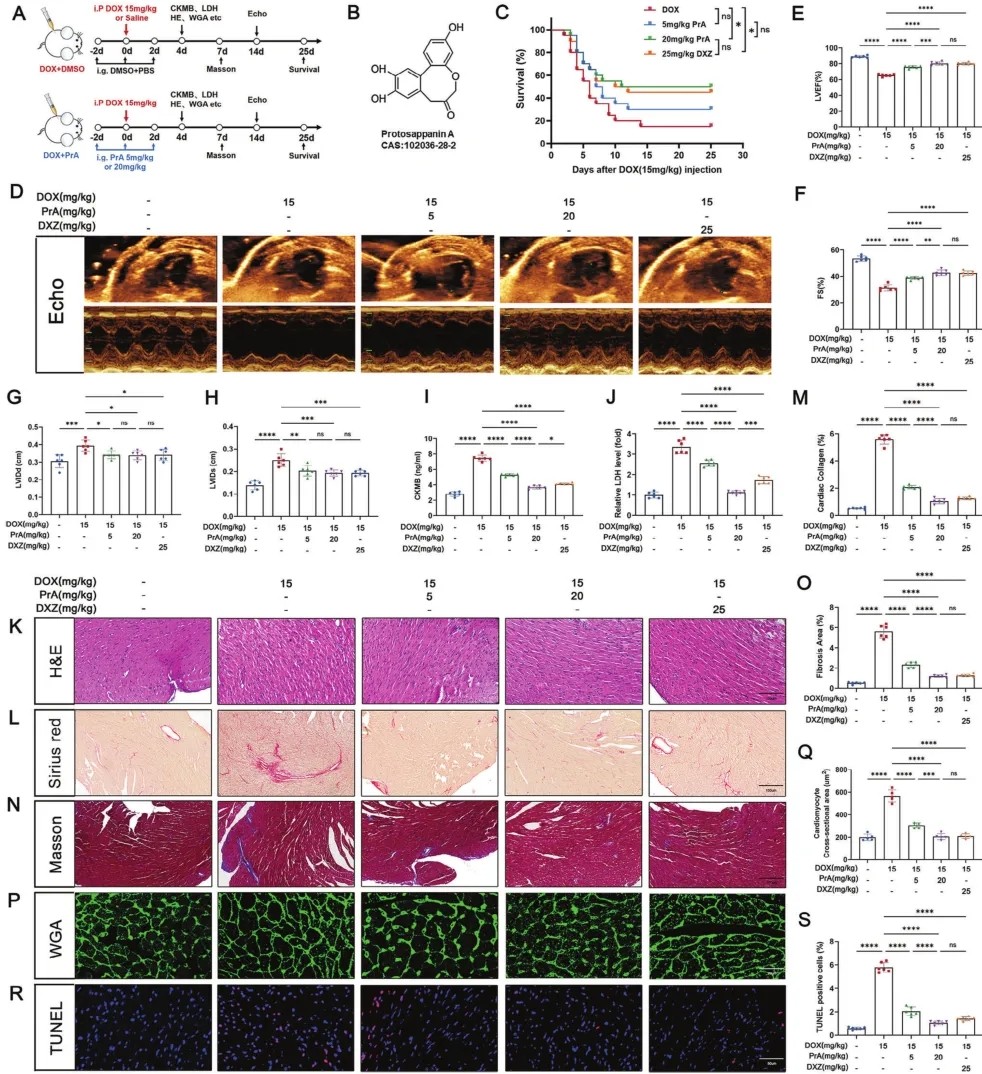
2.PrA抑制DOX诱导的心肌铁死亡
为揭示PrA对DIC保护作用的分子机制,研究人员对三组小鼠(对照组、DOX组和DOX联合PrA处理组)心脏进行RNA测序。对照组的差异表达基因(DEG)(图2A)和京都基因与基因组百科(KEGG)分析(图2B)显示,与对照组相比,DOX条件组中这些DEG中最富集的是铁死亡途径,相反,PrA处理显著降低DOX诱导的与铁死亡相关的DEGs富集(图2C)。此外,基因集富集分析(GSEA)证实,铁死亡被DOX上调,而被PrA下调(图2D),这表明PrA可能通过抑制铁死亡来预防DIC。为验证RNA测序数据,研究人员评估PrA对DIC小鼠铁死亡的影响。分析小鼠心脏中铁死亡标记物Ptsg2的表达采用聚合酶链反应(PCR)。正如预期的那样,PrA剂量依赖性地逆转DOX诱导的Ptsg2基因表达(图2E)。随后,评估小鼠心脏中与铁死亡相关的必需调节蛋白(GPX4、ACSL4和FTH1)的丰度。结果显示,DIC小鼠心脏中ACSL4的含量高于对照组,而FTH1和GPX4的含量则低于对照组。在小鼠中,给药PrA逆转DOX介导的这些蛋白水平的改变(图2F)。免疫荧光进一步支持免疫印迹的发现(图2G)。鉴于铁超载、脂质过氧化和细胞内活性氧(ROS)积累是铁死亡的关键特征,研究人员进一步检测上述条件小鼠中脂质过氧化产物丙二醛(MDA)和谷胱甘肽(GSH)的水平。与对照组相比,DIC小鼠心肌GSH水平降低,心肌和血清MDA水平升高。相反,PrA逆转了DOX诱导的MDA和GSH水平的变化(图2H-J)。此外,DOX给药后心脏组织中总铁和ROS水平显著升高,PrA治疗消除DOX诱导的这些结果(图2K-N)。上述数据表明,PrA负向调节DIC小鼠的心脏铁死亡。
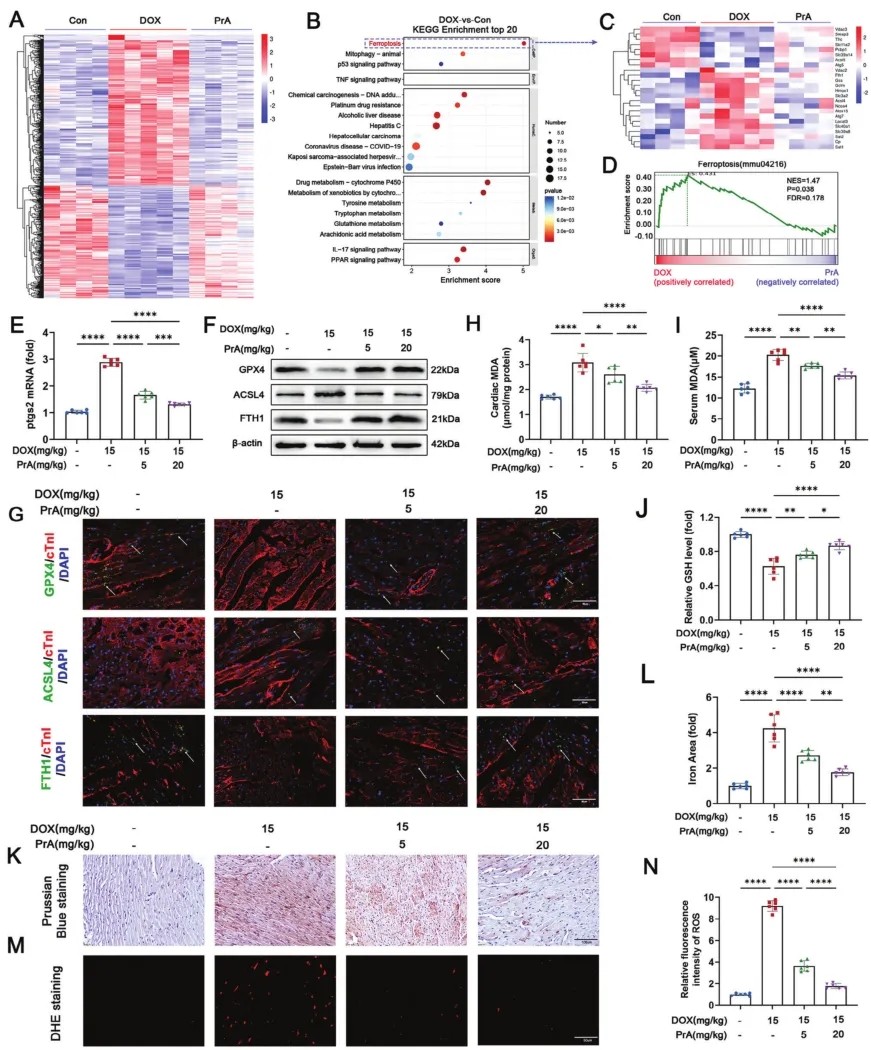
图2 PrA可改善DOX诱导的心脏组织铁死亡。
3.PrA减轻DOX诱导的心肌细胞凋亡并维持线粒体功能
随后,研究人员在体外探讨PrA对大鼠H9C2细胞铁凋亡的细胞保护机制。H9C2细胞用不同浓度的DOX培养24小时后,细胞活力显著降低(图3A)。根据上述细胞活力结果,研究人员选择1×10−6MDOX、50×10−6M和100×10−6MPrA的浓度进行后续实验。根据细胞计数试剂盒8(CCK8)和碘化丙啶(PI)荧光染色的结果,PrA在体外显示出剂量依赖性地减少DOX诱导的心肌细胞损伤,显示出细胞保护作用(图3B-D)。与体内结果一致,PrA处理逆转DOX条件心肌细胞中铁死亡相关参数的水平,包括Ptgs2基因(图3E)、GPX4、ACSL4和FTH1蛋白(图3F)、脂质和细胞内ROS水平(图3G-I)、细胞内铁离子(Fe2+)含量(图3J)、脂质过氧化产物MDA和GSH(图3K,L)。这些结果表明,PrA有效地降低了DOX处理的心肌细胞对铁死亡的体外敏感性。线粒体依赖的铁积累和脂质过氧化是DOX诱导的铁死亡的关键因素。如前所述,研究人员发现在DOX治疗期间,Fe2+和脂质过氧化含量主要在培养的心肌细胞线粒体中增加。然而,PrA处理消除DOX诱导的这种现象(图3N,O)。接下来,进一步监测反映线粒体功能的关键指标,包括线粒体膜电位(ΔΨm)和线粒体衍生ROS(mitoROS)的产生。与对照组相比,DOX处理的心肌细胞ΔΨm减少,线粒体超氧化物积累增加。相反,PrA干预逆转了DOX诱导的这些变化(图3M)。在电镜观察到DOX暴露导致线粒体收缩和线粒体膜密度增加,而PrA处理以剂量依赖的方式改善了DOX介导的小鼠心脏组织异常特征(图3P)。这些结果表明,PrA对DOX诱导的线粒体依赖性铁死亡具有保护作用。
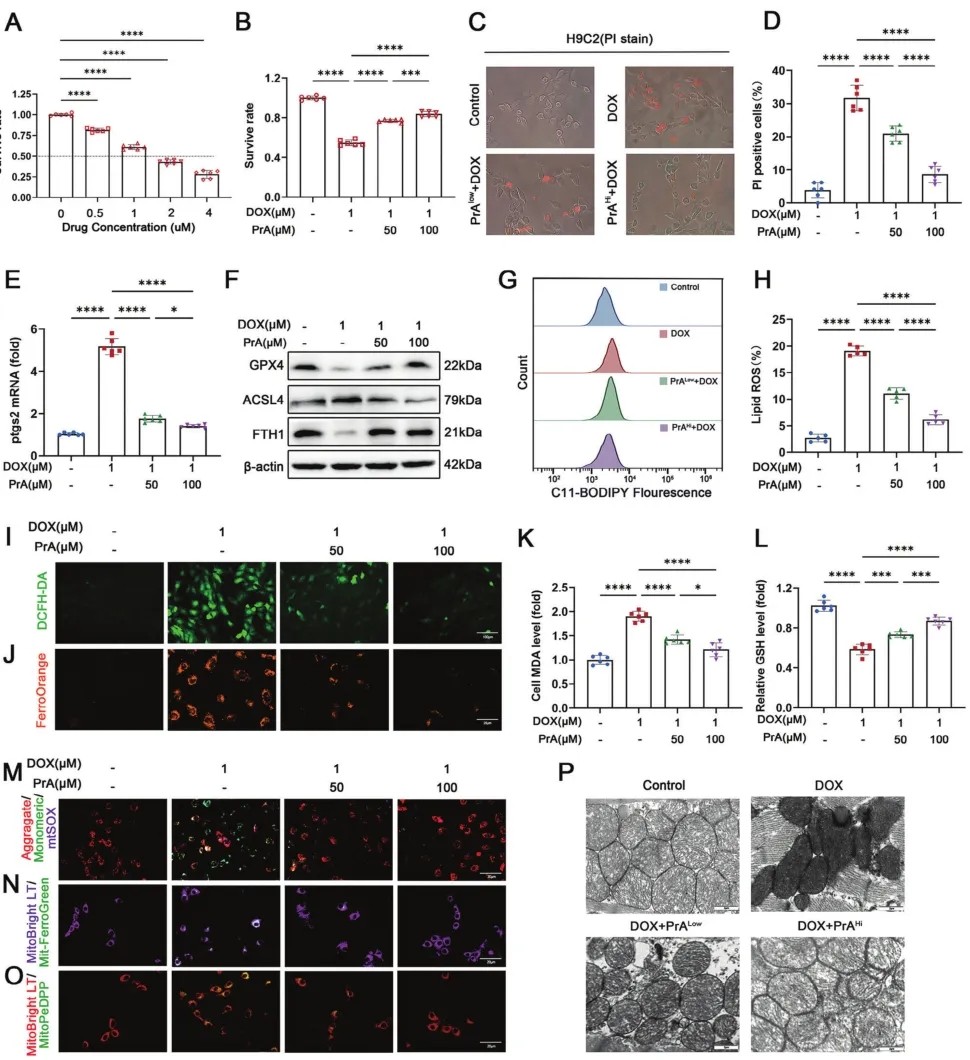
图3 PrA保护H9C2细胞免受DOX触发的铁积累、ROS产生、脂质过氧化,并缓解线粒体功能障碍。
4.ACSL4和FTH1被确定为PrA的直接结合靶点
为确定PrA抑制DOX介导的心肌细胞铁死亡的潜在机制,生物素标记的PrA(Bio-PrA;图4A)与cy5共轭链亲和素(Cy5-SA)串联使用,通过检测PrA与HuProt人蛋白芯片上制备的重组蛋白的结合来识别PrA的潜在靶点(图4B、C)。KEGG分析显示,在PrA结合蛋白组中,铁死亡相关通路富集(图4D)。考虑到PrA在DOX中的重要作用。从z-score中选出参与铁死亡途径的前两个蛋白(ACSL4和FTH1)进行进一步分析(图4E,F)。为验证PrA是否与细胞裂解物中的ACSL4和FTH1结合,Bio-PrA被应用于链亲和素琼脂糖球,并与H9c2细胞的裂解物共孵育。下拉实验显示,Bio-PrA与H9c2细胞裂解物中的ACSL4和FTH1蛋白结合(图4G)。随后,研究人员进行分子对接和动力学模拟,以研究PrA如何与ACSL4和FTH1相互作用。对接结果表明,PrA通过π-π和π-烷基偶联与ACSL4广泛形成疏水相互作用,并通过常规氢键和π-烷基与FTH1表现出亲水性-亲脂性协同作用。所有这些相互作用都有利于稳定结合复合物(图4H)。动力学模拟结果表明,PrA与ACSL4之间平均能量值最高的临界残基为Ile567和Pro445,PrA与FTH1之间平均能量值最高的临界残基为Gln84和Arg23(图4I)。为验证分子对接和动力学模拟结果,研究人员通过细胞热移测定(CETSA)发现PrA与ACSL4和FTH1发生物理相互作用,而在ACSL4的Ile567/Pro445和FTH1的Gln84/Arg23突变后,PrA与ACSL4或FTH1的相互作用明显受损(图4J)。综上所述,PrA直接结合ACSL4和FTH1。
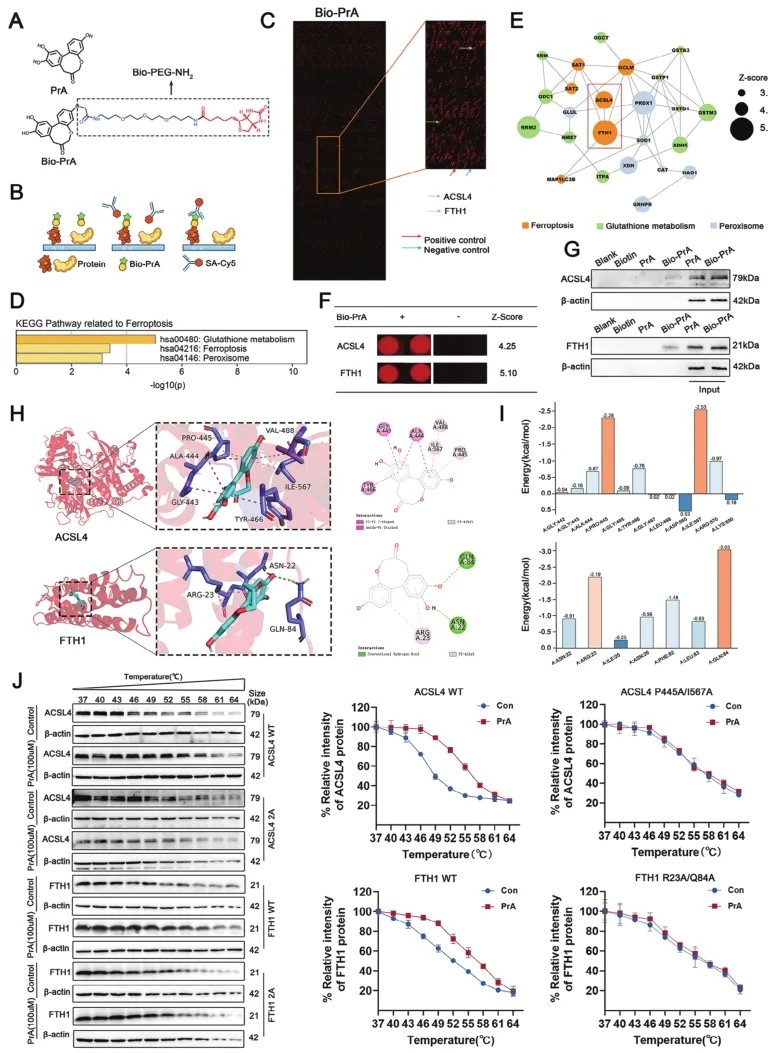
图4 PrA结合蛋白在人蛋白质组芯片上的鉴定。
5.PrA通过阻止ACSL4磷酸化和激活抑制DOX诱导的铁死亡
ACSL4是一种负责代谢多不饱和脂肪酸(PUFA)代谢的同工酶。最近的证据表明,Thr328磷酸化是ACSL4酶激活增强铁死亡敏感性所必需的。考虑到PrA直接结合ACSL4并抑制DOX诱导的铁死亡,研究人员研究PrA是否通过干扰ACSL4的酶激活来保护DOX诱导的铁死亡。Western blotting显示,DOX处理小鼠心脏组织中ACSL4磷酸化和蛋白水平均上调,PrA处理以剂量依赖性方式逆转这些变化(图5A)。在DOX条件下的H9c2细胞中,ACSL4蛋白和Thr328磷酸化水平随着时间的推移逐渐升高(图5B、C)。在DOX暴露24小时后,高剂量PrA处理的H9c2细胞中ACSL4蛋白和Thr328磷酸化水平也受到抑制。值得注意的是,PrA预处理也降低DOX条件下H9c2细胞中ACSL4蛋白水平和Thr328磷酸化水平(图5D)。此外,单独使用PrA治疗不会影响小鼠心脏组织或来自对照组的H9c2细胞中的ACSL4蛋白或Thr328磷酸化水平。因此,PrA与ACSL4的结合在DOX存在下扰乱ACSL4 Thr328位点的磷酸化及其蛋白表达。ACSL4介导的PUFA代谢促进了花生四烯酸(AA)含磷脂的合成和脂质过氧化,最终导致铁死亡。因此,研究人员测量在DOX存在的情况下,用或不用PrA处理的H9c2细胞中的AA水平。酶联免疫吸附试验证实,单独进行PrA预处理对AA的产生没有影响;然而,它逆转H9c2细胞上清中DOX介导的AA生成。尽管在DOX后暴露,高剂量PrA仍保留其挽救DOX引起的AA积累的能力(图5E)。基于这些结果,研究人员得出结论,PrA与ACSL4结合抑制DOX诱导的ACSL4磷酸化和酶活性,进一步破坏PUFAs的下游代谢和脂质过氧化。为证实PrA是否通过阻止ACSL4的磷酸化和激活来抑制DOX诱导的铁死亡,在上述条件下,研究人员在转染siRNA或特异性靶向ACSL4的过表达质粒的H9c2细胞中评估Ptgs2 mRNA水平。与PrA给药数据一致,沉默ACSL4(图5F)抑制DOX条件下H9c2细胞中Ptgs2 mRNA水平,表明ACSL4敲低对DOX诱导的心肌细胞铁死亡有抑制作用(图5G)。此外,沉默ACSL4和PrA对DOX诱导的铁死亡的抑制作用与ACSL4 Thr328磷酸化降低有关(图5H)。相比之下,在DOX暴露的H9c2细胞中,ACSL4过表达(图5I)促进Ptgs2 mRNA(图5J)和ACSL4 Thr328磷酸化水平(图5K)的表达。在ACSL4饱和条件下,PrA对DOX介导的H9c2细胞Ptgs2 mRNA(图5J)、ACSL4蛋白和Thr328磷酸化(图5K)以及脂质和细胞内ROS水平(图5L-O)增加的逆转能力降低。流式细胞术显示ACSL4过表达削弱了PrA对DOX诱导的心肌细胞死亡的保护作用(图5P,Q)。上述数据表明,PrA通过阻止DOX诱导的ACSL4 Thr328磷酸化及其随后的激活来抑制心肌细胞铁死亡。
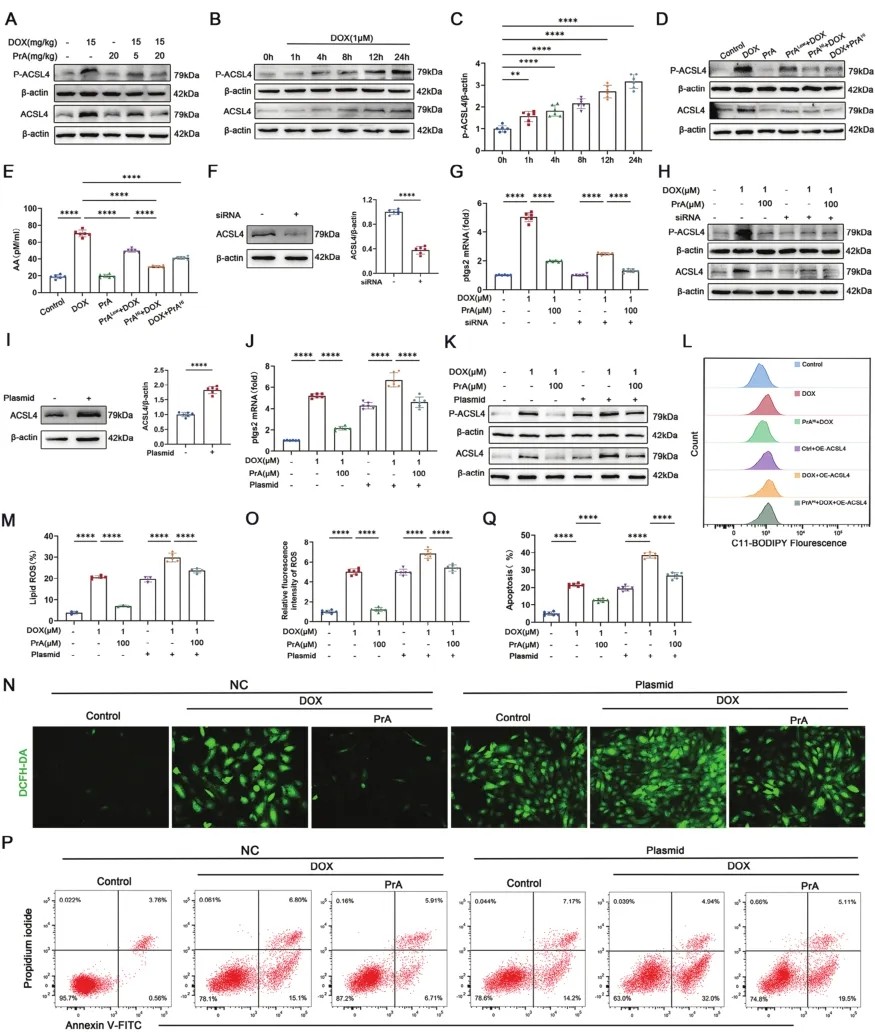
图5 PrA阻断DOX诱导的心肌细胞ACSL4活化,减少ROS生成和脂质过氧化。
6.PrA通过减少溶酶体中FTH1的自噬降解来防止DOX介导的铁凋亡
据报道,铁蛋白的关键亚基FTH1通过结合货物受体NCOA4和转运-介导铁蛋白自噬将含铁的铁蛋白复合物环接在自噬酶体上进行自噬降解,导致游离Fe2+的释放,增加细胞对铁死亡的易感性。之前的数据显示,PrA可以直接与FTH1结合,并在H9c2细胞中恢复DOX诱导的FTH1蛋白水平的改变(图3F)和FTH1蛋白水平在细胞Fe2+生成中的变化(图3J)。因此,研究人员推测PrA可能会破坏DOX介导的NCOA4-FTH1相互作用,随后通过与FTH1结合来干扰铁蛋白自噬。为验证这一可能性,首先使用western blotting分析了使用或不使用PrA处理的DIC小鼠心脏中FTH1、NCOA4和自噬标记物LC3II/I的蛋白表达。与对照组小鼠相比,DOX处理的小鼠心脏组织中FTH1和NCOA4表达较低,LC3-II/I百分比较高,而PrA干预逆转这些变化(图6A-D)。共免疫沉淀实验进一步表明,在DOX处理的小鼠中,给药PrA削弱了FTH1和NCOA4之间的相互作用(图6E)。与体内结果一致,PrA恢复H9c2细胞中FTH1蛋白水平,而不是DOX引起的mRNA表达(图6F),这表明在DOX存在下,PrA通过抑制蛋白质降解而不是上调基因转录来调节FTH1蛋白水平。同样,在DOX暴露前后,PrA挽救H9c2细胞中NCOA4和LC3-II/I百分比的蛋白表达,以及NCOA4-FTH1的相互作用,而单独使用PrA处理对这些指标没有影响(图6G-K)。因此,上述数据支持PrA和FTH1联合抑制DOX触发的NCOA4-FTH1内部相互作用和FTH1蛋白自噬降解的结论。为阐明PrA预防DOX诱导的铁死亡的机制,研究人员使用特异性荧光探针追踪Fe2+和溶酶体,评估细胞内Fe2+的含量和亚细胞位置。激光共聚焦显微镜显示,DOX处理的H9c2细胞在溶酶体中发出强烈的Fe2+荧光,在DOX暴露前增加PrA剂量会减弱这种荧光,这表明PrA抑制DOX诱导的胞内Fe2+从溶酶体释放到细胞质中(图6L,M)。根据免疫荧光分析,DOX处理加速FTH1向溶酶体的易位。相比之下,在PrA诱导下,FTH1和溶酶体的协同作用被取消(图6N)。尽管在DOX治疗后,高剂量PrA仍然抑制FTH1和溶酶体的共定位,以及向细胞质中释放Fe2+(图6L-N)。总的来说,PrA和FTH1的结合破坏NCOA4-FTH1的相互作用,抑制铁蛋白自噬和溶酶体释放铁2+,从而改善DOX诱导的心肌细胞铁凋亡。
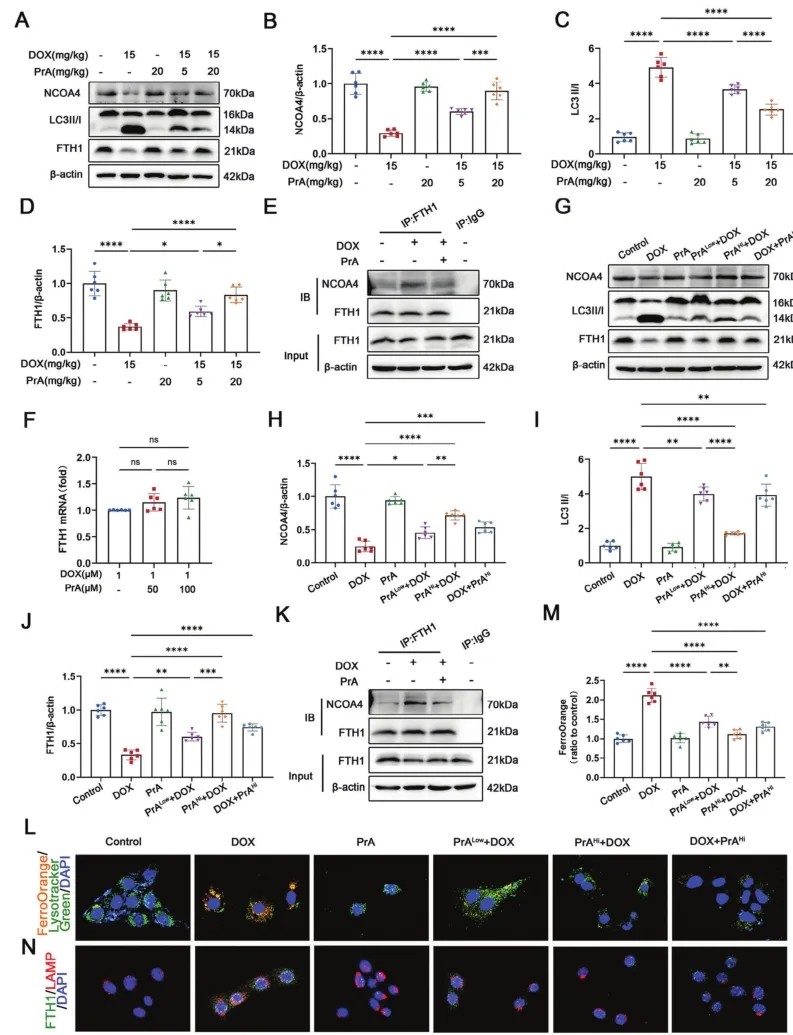
图6 PrA抑制FTH1在溶酶体中的自噬降解。
7.PrA介导的铁死亡抑制减轻缺血再灌注诱导的心脏损伤和功能障碍
最近的研究表明,铁死亡与心肌I/R之间存在很强的相关性,表明铁死亡干预可能在再灌注后增强心功能方面具有治疗益处。因此,研究人员研究PrA治疗是否对心肌I/R损伤有治疗作用。建立心肌I/R小鼠模型,给予或不给予PrA治疗指定天数(图7A),与I/R小鼠相比,PrA治疗部分恢复I/R诱导的血清心肌损伤标志物活性升高,包括CK-MB和LDH(图7B,C)。PrA预处理组梗死面积明显小于I/R组(图7D)。随后,使用超声心动图评估心功能。I/R诱导的心功能障碍主要表现为LVEF、FS、LVIDd和LVIDs的变化,经PrA治疗后恢复(图7E-I)。此外,组织学染色显示,PrA显著改善I/R诱导的心脏纤维化(图7J)。为阐明PrA对心肌I/R损伤中铁死亡的保护机制,研究人员采用PCR方法检测I/R小鼠心脏中Ptsg2的mRNA水平。正如预期的那样,PrA处理的I/R小鼠Ptgs2 mRNA水平显著下调(图7K)。此外,I/R组心脏和血清MDA水平高于PrA治疗组(图7L,M)。PrA处理恢复I/R诱导的小鼠心脏组织中GPX4、ACSL4和FTH1蛋白(图7N)、铁(图7O)和ROS含量(图7P)的变化。上述研究结果表明,PrA对心肌I/R损伤具有抗心肌收缩和心脏保护作用。
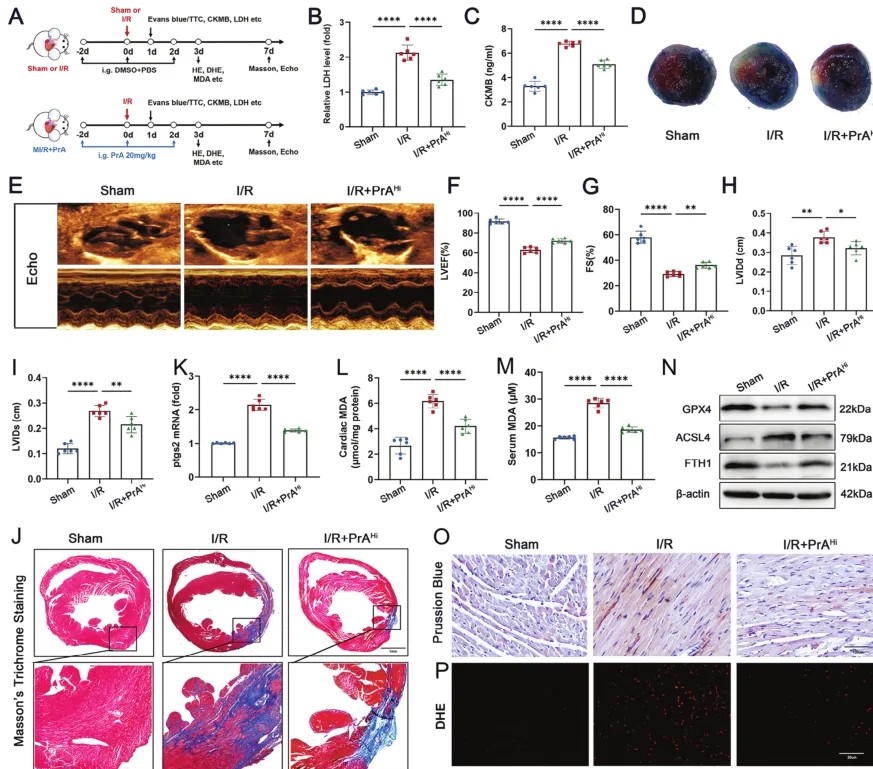
图7 PrA可改善缺血/再灌注(I/R)诱导的心脏损伤和铁死亡
文章小结
本研究揭示PrA通过靶向ACSL4/FTH1轴依赖的铁死亡途径,对DOX诱导的心肌损伤和心脏功能障碍具有保护作用。此外,PrA能够减少DOX诱导的心肌细胞铁死亡,维持线粒体功能,并在I/R损伤模型中显示出心脏保护效果。研究数据明确显示,PrA具备作为一种新型双重铁死亡靶向抑制剂的潜力,其在DIC以及其他铁死亡介导的疾病的治疗途径中展现出有希望的进展和转化价值。看完这篇文章有没有给你带来一丝灵感?没有idea也没关系,扫码联系小仲,让小仲帮你解决idea问题。小仲这里可以提供各种优质服务,主打就是一个满意!
